> ## Documentation Index
> Fetch the complete documentation index at: https://docs.cosmosid.com/llms.txt
> Use this file to discover all available pages before exploring further.
# Uploading Sequencing Data
> The Cosmos-Hub supports importing raw FASTQ data for both amplicon and shotgun metagenomic workflows using multiple upload sources.
**Samples are analyzed on a per-sample basis**, and uploading requires pre-purchased sample credits. Contact [info@cosmos-hub.com](mailto:info@cosmos-hub.com) for pricing.
# There are 4 upload methods to choose from:
Upload local FASTQ, SRA, or BAM files via drag-and-drop.
Connect your Illumina BaseSpace account and import data directly.
Use public SRA accession numbers to fetch data.
Use our CLI or API for large batch uploads and automation.
(Click for API documentation)
***
# Select Your Data Type
Amplicon pipelines are used to identify bacterial or fungal communities based on specific genes. These workflows support both ****single-end and paired-end**** reads.
* **Read type**: Paired-end or single-end
* **Min samples**: 10 (for ASV/DADA2 workflows)
* **File size**: ≤ 1 GB uncompressed (250 MB compressed)
* **PHRED score** ≥ 20
* **Formats**: `.fastq.gz`, `.fq.gz`, `.fasta.gz`, `.sra.gz`
* **Paired-end naming**:
* `SampleID_R1_001.fastq.gz`
* `SampleID_R2_001.fastq.gz`
* Reads must **overlap ≥ 15 bp**
* **Watch the 16S Tutorial Video**
* **ASV 16S rRNA Profiling** (DADA2-based)
* **OTU ITS rRNA Profiling** (QIIME2-based)
* Filenames with **spaces or special characters** will break pairing
* Unpaired files will appear as "Single-read samples"
Shotgun workflows analyze entire microbial genomes and functional markers. These require **paired-end** reads and support large files.
* **Read type**: Paired-end only
* **File size**: ≤ 50 GB
* **PHRED score** ≥ 20
* **Formats**: `.fastq.gz`, `.bam`, `.fq.gz`, `.sra.gz`
* **Naming**:
* `SampleID_R1.fastq.gz`
* `SampleID_R2.fastq.gz`
* Select host genome (e.g., Human, Mouse, Pig)
* If using **CHAMP**, select **"None"** — it performs built-in host filtering
* **CHAMP**: Human-specific taxonomic & functional profiling
* **Kepler**: Host-agnostic taxonomic profiling
* **Host-Agnostic Functional Profiling**: MetaCyc, GO, EC, Pfam, CAZy
* **AMR/Virulence Marker Detection** *(requires add-on module)*
The [Sequence Read Archive (SRA)](https://www.ncbi.nlm.nih.gov/sra) is a publicly accessible database of raw sequencing data hosted NCBI. It contains high-throughput sequencing data from numerous research projects worldwide, covering a wide range of study types and organisms, including metagenomic and microbiome studies. Researchers deposit their sequencing reads in SRA to make data publicly available in accordance with scientific publication practices, fostering transparency and reproducibility in science. This is typically referenced in the paper with a [BioProject](https://www.ncbi.nlm.nih.gov/bioproject) number in the Data Availability section.
**Why Import SRA Data for Microbiome Analysis?**
**Access to Publicly Available Data**: Researchers can leverage publicly available SRA datasets to replicate studies, validate findings, or perform meta-analyses against their own data.
**Cost Efficiency**: Using existing SRA datasets reduces the need for sequencing of new samples when performing secondary analyses or meta-analyses. By importing previously published data, users can rapidly evaluate different datasets or integrate multiple studies without the need to collect and sequence new samples.
**Extended Contextual Analysis**: By analyzing previously published microbiome datasets from SRA, researchers can add context to their own data. This can provide valuable insights by comparing microbiomes across different populations, environments, diseases, or experimental conditions.
**Data Reuse for Novel Insights**: Many research questions can be answered by revisiting older datasets with new hypotheses or more advanced analysis techniques. The Cosmos-Hub allows users to explore data from fresh perspectives using its comprehensive suite of microbiome analysis tools, potentially leading to novel discoveries.
**Be sure to leverage the SRA Run Selector to export tables of SRA accession IDs as well as relevant metadata. Typically, original manuscripts need to be referenced for primer sequences.**
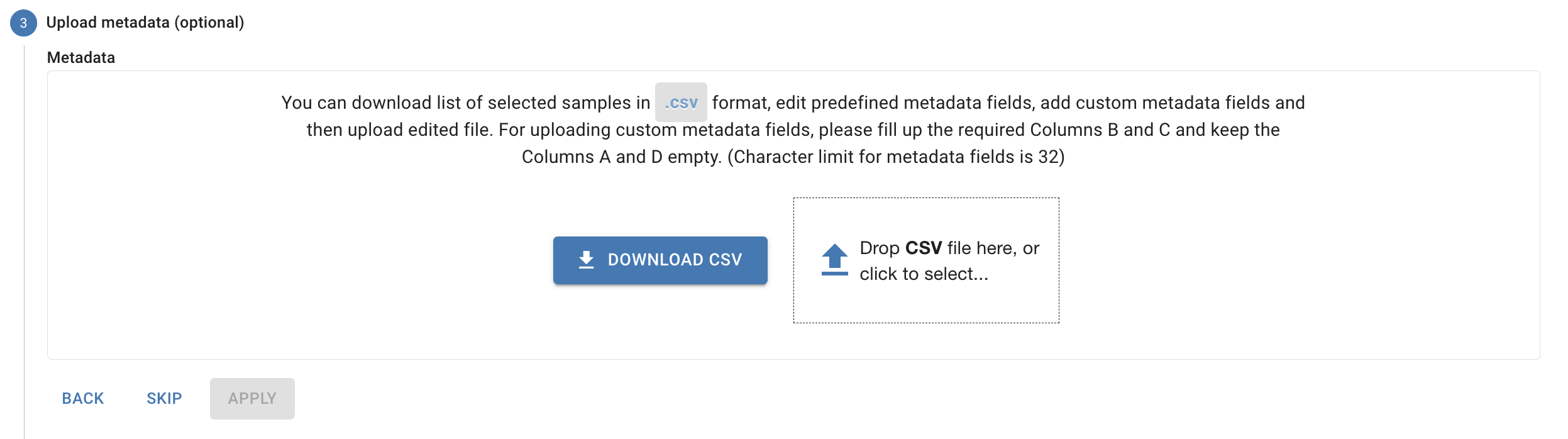
# Uploading Metadata
You can upload a metadata CSV during or after your data upload through the **Cohorts and Metadata** page.
***
# Workflow Selection
After upload, choose a workflow that matches your data type. The Cosmos-Hub will only show valid options based on your selections.
***
**Do not close your browser while uploading.** Progress is shown in the bottom-right. Allow up to 24 hours for processing. Results and statuses will appear in the **Cohorts and Metadata** section.
Occasionally a sample will fail to upload. Here are a number of possible causes:
* Poor internet connection
* Internet connection interruption
* Browser closed while uploading
* Incorrect primer pairs
* Corrupted or low quality data files
Other notes:
* If a sample fails to upload, you can retry it
* If a sample is not in the proper format or fails to run for another reason, the status will show “failed” and you will not be charged credits.
* If you have problems uploading or with a failed sample, [please submit a help ticket](www.cosmos-hub.com/support) or email us at [help@cosmos-hub.com](mailto:help@cosmos-hub.com).
 ***
# Workflow Selection
After upload, choose a workflow that matches your data type. The Cosmos-Hub will only show valid options based on your selections.
***
# Workflow Selection
After upload, choose a workflow that matches your data type. The Cosmos-Hub will only show valid options based on your selections.
 ***
***