Metadata Upload Improvements
Metadata Upload Enhancements
We have made several improvements to the metadata upload process in Cosmos-Hub:- Transposed Metadata Sheet Support: The platform now accepts both long format (default) and short format (transposed) metadata templates, giving you flexibility in how you organize your data. See the Uploading Metadata page for details on both formats.
- Improved Error Handling: Enhanced error messages provide clearer guidance when metadata uploads encounter issues, helping you quickly identify and resolve problems with your files.
- Better User Experience: Streamlined upload workflow with more intuitive feedback throughout the metadata import process.
RITA
Annoucing RITA, your AI Microbiome Assistant
We are excited to introduce and expand RITA (Revealing Insight Through Ai), our intelligent AI assistant for microbiome data interpretation. RITA is built to enhance and accelerate the research experience for Cosmos-Hub users with these capabilities:- Comprehensive Literature Integration: curated database of microbiome/microbiology scientific literature, public databases, and Cosmos-Hub’s proprietary knowledge base
- Natural Language Query Support: Users can ask RITA a wide range of questions, from details about microbial taxa, gene functions, and biological pathways to disease associations. RITA can also analyze and interpret uploaded datasets, offering insight directly from study results.
- User-Friendly Access: RITA is accessible via a dedicated web chat, designed for both simple and complex inquiry. Account setup is quick with self-service registration and activation support.
- Research-Only Guidance: RITA is intended for research and informational purposes, supporting R&D and knowledge generation. For clinical use cases, users are reminded to consult qualified professionals.
- Continuous Improvements: The RITA knowledge base is regularly updated, ensuring access to the latest research findings and microbiome data interpretation capabilities.
Data Sharing
Data Sharing Updates
We are pleased to announce enhancements to our Data Sharing feature, empowering collaboration across teams and organizations:- Team Access for Annual Subscribers: Data sharing is now available for users with active annual subscriptions. This feature enables secure sharing of data and results between multiple accounts, typically within a research team or organization.
- Streamlined User Management: To add additional users or teammates to your sharing network, customers can contact Cosmos-Hub support for prompt activation and help configuring access.
- Study Dashboard Integration: Shared data, including raw sequencing files, profiling results, and metadata, are linked seamlessly within the Cosmos-Hub study dashboards. Users benefit from integrated cloud storage, making multi-user collaboration and secure data access effortless.
- Security and Privacy: Data sharing operates on secure AWS infrastructure, with institutional account management, unique credentials, and multi-factor authentication to protect sensitive research data.
- Support and Onboarding: For setup help or troubleshooting, Cosmos-Hub support is available to ensure teams can maximize the value of shared resources.
Module Release: Advanced Statistics
Advanced Statistics Module now available
We are thrilled to introduce the Advanced Statistics Module to CosmosID-HUB, offering even greater analytical power for your microbiome research! This exciting feature is built on the robust MaAsLin2 framework and includes early access to MaAsLin3 (Beta), taking your data analysis to the next level.This exciting new module will enable users to leverage their study metadata even further and run more complex statistics with their microbiome profiles.The Advanced Statistics Module enables users to:
- Leverage continuous variables for correlative microbiome analysis
- Use multivariate statistics to control for potential confounders
- Use time series data to perform longitudinal analysis
- Customize their statistical analysis with various parameters and metrics
- Generate new and dedicated visualizations
Pipeline Release: CHAMP™ Human Microbiome Profiler
CHAMP Human Microbiome Profiler is now available in the CosmosID-HUB
- CHAMP offers high-resolution taxonomic and functional profiling that is unparalleled in accuracy, precision, and coverage to maximize insights from human microbiome studies.
- CHAMP™ employs over 400,000 metagenome-assembled genomes (MAGs), created from a collection of more than 30,000 human microbiome samples across 9 different body sites from individuals across the world.
- Enables detection of nearly 7000 bacteria, archaea, and eukaryote species, including many of which are newly discovered.
- Functional characterization via Gut Metabolomic Module (GMM), Gut Brain Module (GBM), KEGG Modules & Orthologs (requires license)
Why CHAMP™?
- Improved Sensitivity and reduced risk of missing important species
- Reduced false signal and increased confidence in detection
- Higher resolution and fewer unknown species via GTDB nomenclature
Read the paper and benchmarking stats in Frontiers of Microbiology (Pita et al, 2024)
New Subscription Model for the CosmosID-HUB
CosmosID-HUB is now subscription based
The CosmosID-HUB is moving to a more traditional ‘Software-as-a-Service’ (SaaS) model based on an annual license/subscription. With this change, HUB users will benefit from:- Decreased sequencing pricing, effective as of today.
- More inclusive profiling - shotgun profiling will now include taxonomy and function together, as standard, giving you access to over 10 different databases with a HUB subscription.
- Access to licensable databases such as KEGG, VFDB, CARD etc.
When will it change?
For our existing customers who wish to continue to use the software, we are aiming to have everyone transitioned over by July 1st, 2024.What if I don’t want to enter into a HUB subscription?
Whilst we’d hate to see you go, you are of course entitled to all of your profiling data, which we’d ask you to go ahead and download for your records before July 1st, 2024.Why is this changing?
Ultimately, your investment into the HUB software enables us to accelerate its development and take your research even further. Some tools and functionalities that we’re working on include:- CHAMP - our next-generation human microbiome profiler
- An updated Virulence Factor database
- A KEGG module, including licensing access
- Advanced Statistics
- An Improved User Interface
- Visualizing data from external pipelines
- A Metabolomics module
What do I do now?
This change in service means that we will be able to offer you an even wider range of functionalities and tools that benefit your projects. Would you like a demo of the latest capabilities of the platform or have any questions? Our team is ready to assist. Your research journey is important to us, and we’re committed to ensuring it’s as impactful as possible.Database Release: Bacterial/Fungal
Bacterial/Fungal Database Update
We are excited to release updated bacterial and fungal databases with > 15,000 new curated genomes, bringing our total to more than 170,000 microbial genomes and gene sequences representing bacteria, viruses, protists, and fungi, as well as antibiotic resistant and virulence associated genes. With thousands of new strains and species, we are now able to characterize complex microbiome communities with increased sensitivity and specificity.The CosmosID databases are organized phylogenetically and contain hundreds of millions of marker gene sequences. The markers represent both coding and non-coding sequences uniquely identified by taxon and/or distinct nodes of phylogenetic trees. This means that the tree structure was created based on genomic relatedness of organisms rather than predetermined taxonomy based on phenotype. This allows CosmosID to have a high degree of accuracy in identifying microorganisms based on their DNA in metagenomic samples. It also helps identify the closest match to genomes that do not have strain level references in the database (if, for example, they have never been sequenced before).We have now switched from NCBI- to GTDB-based nomenclature.
Classifying bacteria is crucial for understanding their ecology, evolution, and potential roles in health and disease. Two prominent systems for bacterial taxonomy are the National Center for Biotechnology Information (NCBI) taxonomy and the Genome Taxonomy Database (GTDB). While both aim to categorize bacteria, they differ in their underlying philosophies and methodologies, leading to discrepancies in classification.The NCBI taxonomy, a long-standing system, curates classifications based on a combination of phenotypic and genotypic data [1]. However, in recent years, researchers highlighted inconsistencies arising from historical practices and the subjective nature of phenotypic traits [2]. Additionally, the NCBI taxonomy is not strictly rank-normalized, meaning equivalent evolutionary distances can be assigned different taxonomic ranks [1].GTDB, a newer system, focuses on a phylogenetically consistent and rank-normalized classification based on whole-genome sequences. It utilizes Average Nucleotide Identity (ANI) to delineate species boundaries and Relative Evolutionary Divergence (RED) to define higher taxonomic ranks [2]. This approach aims to provide a more objective and evolutionarily informative classification scheme leading to GTDB often recognizing finer taxonomic levels and proposing novel lineages not present in the NCBI taxonomy . Here’s a breakdown of the key differences between GTDB and NCBI taxonomies:- Philosophy: NCBI - Curator-driven, phenotypic and genotypic data; GTDB - Genome-based, phylogenetically consistent.
- Data source: NCBI Refseq and Genbank; GTDB - NCBI Refseq and Genbank
- Species definition: NCBI - Less stringent, often based on 16S rRNA gene sequence similarity [1]; GTDB - Stricter, based on ANI values (>95% for bacteria) [2].
- Higher-rank classification: NCBI - Traditional Linnaean ranks (phylum, class, order, etc.); GTDB - Ranks are normalized based on evolutionary divergence (phylum level for Bacillota can be Bacillota, Bacillota_A, Bacillota_B; ) [2].
- Schoch, C.L., Ciufo, S., Domrachev, M., Hotton, C.L., Kannan, S., Khovanskaya, R., Leipe, D., McVeigh, R., O’Neill, K., Robbertse, B., Sharma, S., Soussov, V., Sullivan, J.P., Sun, L., Turner, S. and Karsch-Mizrachi, I., 2020. NCBI Taxonomy: a comprehensive update on curation, resources and tools. Database, 2020, baaa062.
- Parks, D.H., Chuvochina, M., Waite, D.W., Rinke, C., Skarshewski, A., Chaumeil, P.-A., and Hugenholtz, P., 2018. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nature Biotechnology, 36(10), pp.996-1004
Pipeline Release: Kepler Host-Agnostic Microbiome Profiler
Kepler™ Host-Agnostic Microbiome Profiler
Kepler™ uses a host-agnostic curated database to process samples beyond the human host including but not limited to environmental, animal, soil, food samples and many others.Kepler extracts optimal value from metagenomic data by combining the precision of K-mer exact-matching and the versatility of probabilistic alignment. Through this method, Kepler achieves robust identification and enumeration of bacteria, viruses, fungi, and protists by leveraging a meticulously curated biomarker database, where over 30,000 species are arranged in a phylogenetic tree-like structure.The core of Kepler’s technology is patented in both the US Patent office (US10108778B2, US20200294628A1) and European Patent Office (ES2899879T3).Most notably, Kepler marks a transition from NCBI-based nomenclature to GTDB-based nomenclature. Read more about [the NCBI vs. GTDB transition](GTDB vs. NCBI Taxonomic Nomenclature).Kepler Advantages
How does Kepler work?
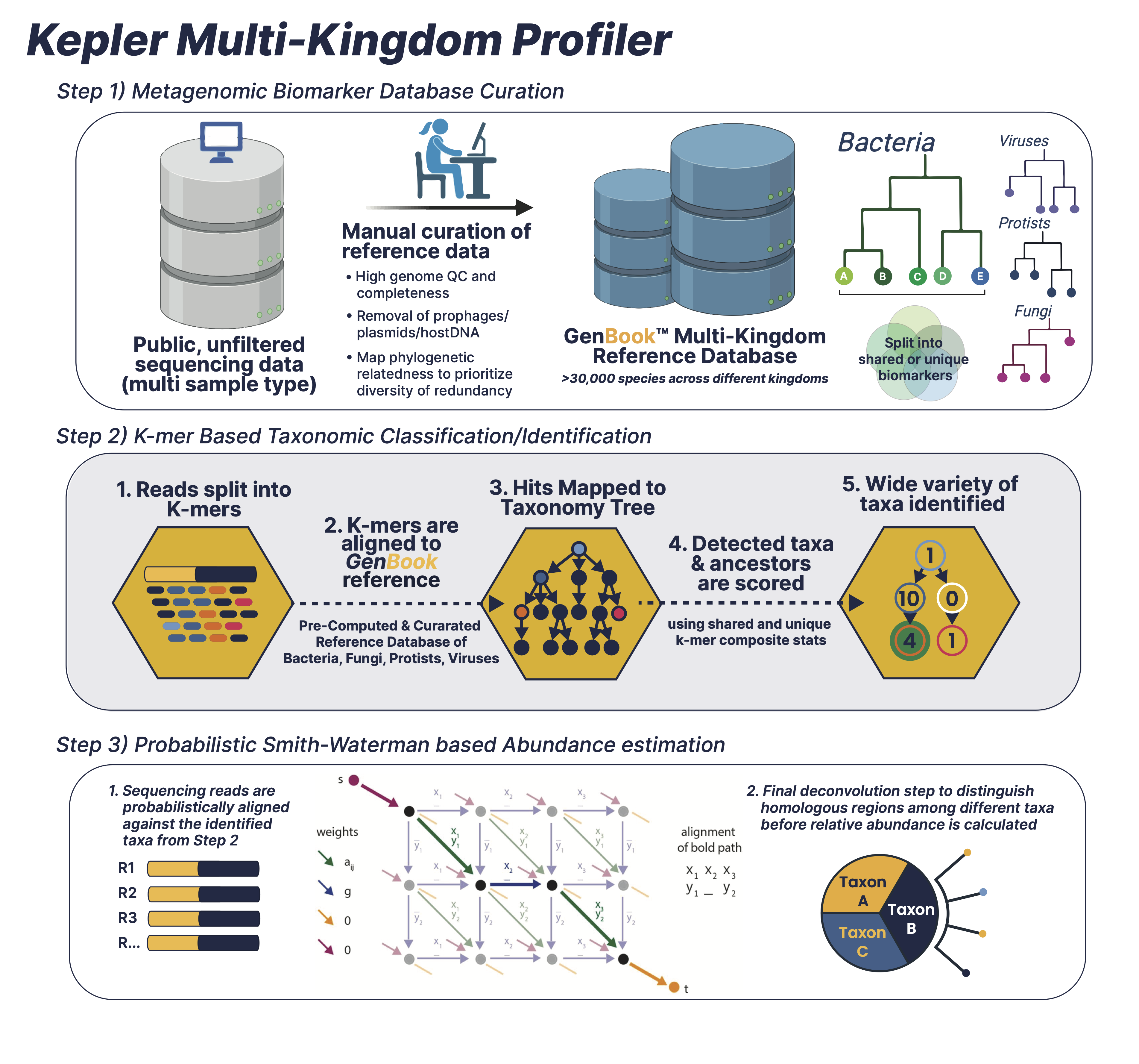
1. Leveraging a Curated Database of Microbial Genomes
The Kepler database of high quality microbial genomes is based on high completeness:low contamination ratio, genome assembly quality and prioritizing intra-species diversity whilst limiting phylogenetic redundancy. The genome assemblies are then scrubbed clean of low complexity sequences, prophages, plasmids and host-contaminated regions to maximize the taxonomic signal-to-noise ratio. The final database encompasses multiple microbial kingdoms and >30,000 species.2. Identifying Relevant Biomarkers
With the genomes curated and cleaned, they undergo a pre-computation phase where they’re split into n-mers of variable length. The n-mers are then categorized as either shared or unique biomarkers across individual genomes, which is facilitated by a phylogenetic tree-like data structure. The tree backbone represents shared genomic biomarkers between different taxa, while the tree leaves are individual microbial genomes with unique biomarkers.3. Searching the Biomarker Database
The second per-sample, computational phase searches the millions of short sequence reads or contigs in your data against the phylogenetic tree-like database build:- The first comparator splits the sequencing reads into k-mer sets that are then queried across the different branches and leaves of the phylogenetic tree to identify the different taxa present in the query kmer-sets. The first comparator splits the sequencing reads into k-mer sets that are then queried across the different branches and leaves of the phylogenetic tree to identify the different taxa present in the query kmer-sets. The first comparator looks for exact matches between query k-mers and reference bio-markers and classification sensitivity and accuracy is maintained through composite k-mer/biomarker aggregation statistics and coverage depth estimation.
- The second comparator uses an edit distance-scoring based probabilistic Smith-Waterman algorithm to compare sequencing reads with a reference set of identified microbial taxa using the first comparator. In conclusion, overall abundance precision and classification accuracy is achieved by running the comparators in sequence, scoring the entire read probabilistically against the reference set, and a final deconvolution step to distinguish homologous regions.
Evaluation of Kepler with Biological Community Standards
To benchmark Kepler, real-world community standards were utilized to compare its efficacy against leading profilers such as Kraken2/Bracken and MetaPhlAn4. For these comparisons, 5 different community standards were employed with both even and staggered (log distribution), from ATCC and Zymo.Kepler distinguished itself not only by achieving a superior F1-Score (a balanced measure of precision and sensitivity) but also by its exceptional ability to detect low-abundance taxa (Bacteria and Fungi) as well as its precision in differentiating closely related taxa at the sub-species level, for example, Bifidobacterium longum subsp. longum and Bifidobacterium longum subsp. infantis.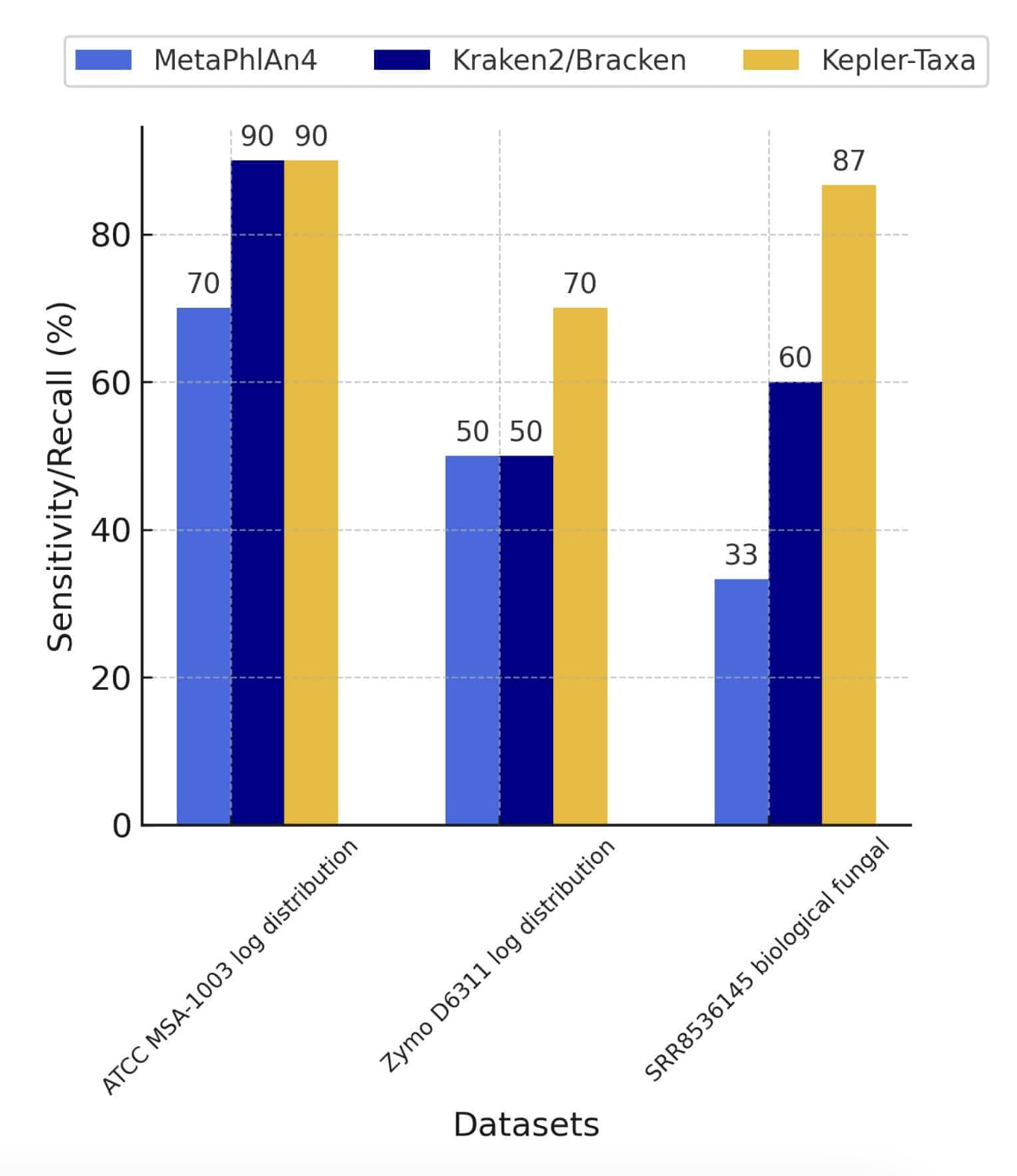
Figure 1: This figure illustrates the sensitivity of Kepler in comparison to other methods when applied to staggered community standards and fungal biological dataset. Kepler significantly outperforms in detecting both Bacteria and Fungi within these staggered standards, showcasing its superior sensitivity.
Pipeline Release: Host-Agnostic Functional Workflow
Host-Agnostic Functional Workflow
This year, we are pleased to announce two major updates to our functional profiling workflow (Functional v1.0 → 2.0). One of the primary focuses of this latest release was to improve the functional characterization of the microbiome by expanding our catalog of gene families from Uniref90. The update to the Uniref90 gene family database has substantially increased our detection of gene functions in the microbiome community, which can be seen in Figure 1 with an increase in the number of GO Terms detected in both human stool and mouse gastrointestinal tract specimens.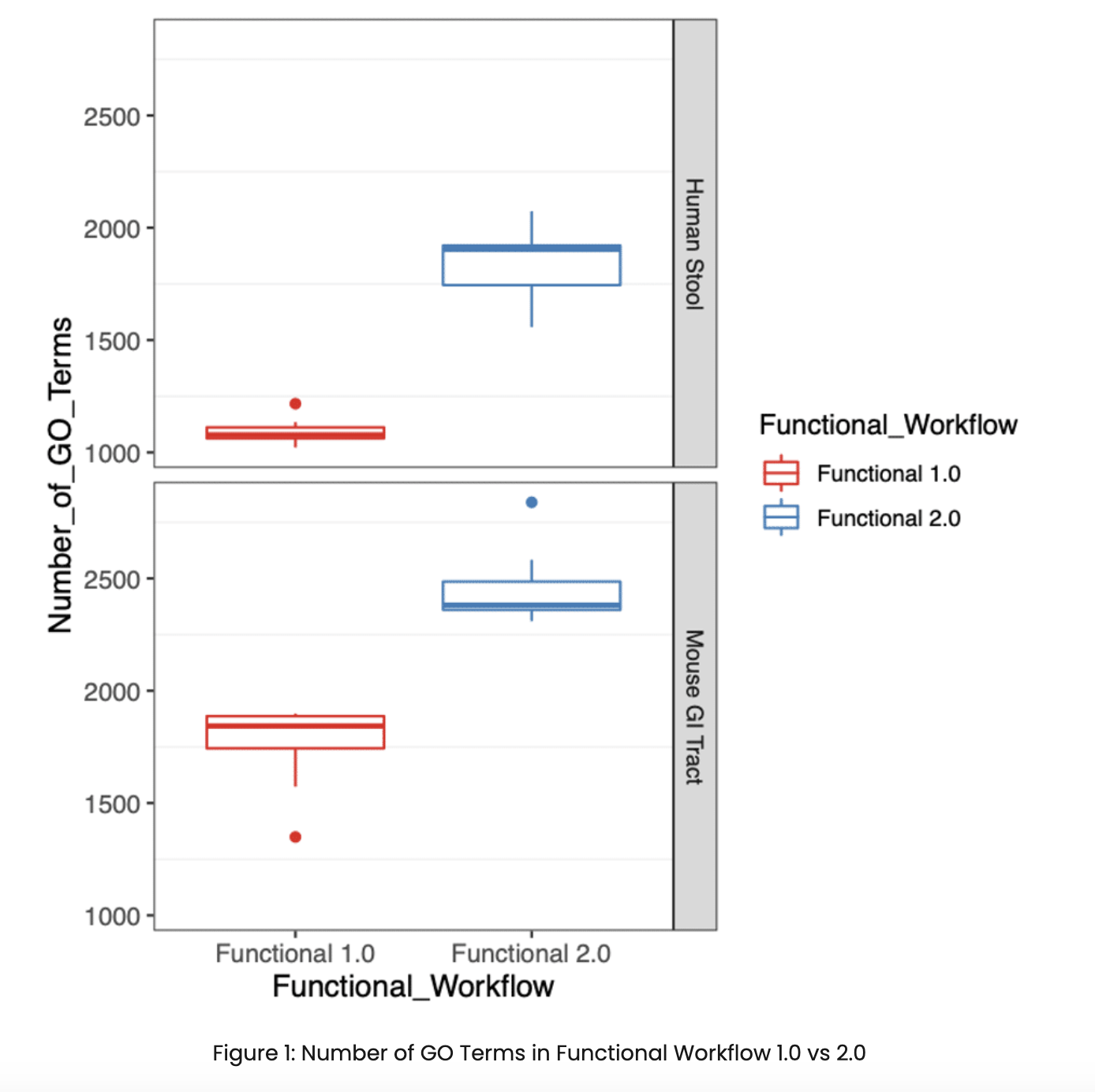
- Enzyme Commission Database: a database of the numerical classification of enzymes based on the reactions they catalyze
- Pfam Database: a database of protein families generated using hidden Markov models
- CAZy Database: a database of structurally related catalytic and carbohydrate-binding modules of enzymes that degrade, modify, or create glycosidic bonds