CHAMP is published in Frontiers in Microbiology! Read about pipeline and benchmarking stats in our recent paper, “CHAMP delivers accurate taxonomic profiles of the prokaryotes, eukaryotes, and bacteriophages in the human microbiome”
Overview
CHAMP is a next-generation human microbiome profiling pipeline developed to deliver high-resolution taxonomic and functional insights from shotgun metagenomic sequencing data. By leveraging an expansive and highly curated reference database, CHAMP offers precise species-level identification, robust functional potential profiling, and superior performance compared to other commonly used tools in the field. Some highlights of the pipeline include:- Species-level resolution across prokaryotes, eukaryotes, and viruses.
- Built on 400,000+ metagenome-assembled genomes (MAGs) from >30,000 human microbiome samples
- Benchmark-leading accuracy compared to MetaPhlAn4, Kraken2, Bracken, and Centrifuge
- Functional annotations from Gut-Brain Module (GBM), Gut-Metabolic Module (GMM), and KEGG
- Supports phage/virome detection, strain-level profiling, and clonal resolution (custom analysis; contact info@cmbio.io)
.png?fit=max&auto=format&n=1_URKzklO7hYjRsk&q=85&s=6faae1115d215530cbd5516dd2f97702 "CHAM Pinfographic(minusviralgenomes) Pn")
Use Cases:
- Human health studies (e.g., IBD, metabolic disorders)
- Probiotic development (strain-specific tracking, clonal resolution)
- Population-scale microbiome research
- Virome and bacteriophage profiling
Reference Database Construction
- Reference catalog is derived from:
- Publicly available MAGs (UHGG, ELGG)
- Clinical Microbiomics/Cmbio in-house MAGs
- NCBI/PATRIC genomes (to capture pathogens, probiotics, food-related species)
- MAG Processing
- Assembly tools: Megahit, metaSPAdes
- Binning: VAMB
- Quality control: CheckM2 (>90% completeness, <5% contamination), GUNC
- Annotation: GTDB-Tk with GTDB r214
- Species Clustering
- MAGs clustered at 95% ANI using dRep/FastANI
- Pan-genomes constructed in 3 stages using MMseqs2 and CD-HIT
- Final database includes ~6,567 prokaryotic species clusters and 244 eukaryotic species
- Final non-redundant gene catalog: >25 million genes
Species Quantification
Signature Gene Selection Each species is represented by up to 250 signature genes, selected for:- Uniqueness (no cross-species matches at >97% identity for >=100 bp)
- Core gene status (present in >=60% of MAGs)
- Length (>=200 bp and <=20 kbp)
Read Mapping & Quantification
- Preprocessing:
- Human host-filtering (GRCh38 via Bowtie2)
- Adapter and quality trimming (AdapterRemoval)
- Paired-read retention (both reads >=100 bp)
- Read Mapping:
- BWA-MEM with >=95% identity over >=100 bp and MAPQ >=20
- Reads categorized as uniquely mapped, multi-mapped, or unmapped
- Abundance Estimation:
- Based on a negative binomial model using signature gene counts
- Adjusted for gene length and mapping confidence
- Outlier filtering via quantile boundaries
- Final species abundances normalized to sum to 100% per sample
Read Count Adjustment and Reporting
Due to the sample-specific removal of outlier genes from abundance estimation, effective genome length varies across samples. As a result, raw species read counts are not directly proportional to estimated species abundance. To correct for this, raw species read counts are adjusted to reflect abundance using the following procedure:- Each species abundance is multiplied by the sum of effective gene lengths for its signature genes (all signature genes are included in the sum, even those flagged as outliers).
- These scaled values are then normalized so that their sum matches the total number of reads mapping to signature genes in the sample.
- Finally, adjusted values are rounded to integers, minimizing rounding discrepancy and ensuring the aggregate read count remains unchanged
**Why are counts and RA (Relative Abundance) not proportional?
**Species read counts and relative abundance (RA) are not directly proportional because outlier signature genes are removed per sample, causing the effective genome length of each species to fluctuate. This technical adjustment ensures consistency but means that counts and RA reflect different underlying calculations, as described in the methodology above
Functional Profiling
Functional Annotation- Prokaryotic genes: Annotated using EggNOG-mapper (v5.0) for orthologous groups and KEGG Orthology (KO)
- Eukaryotic genes: Annotated with KofamScan using KO assignments
- KEGG Modules
- Defined as sets of KO identifiers encoding a biological function or pathway
- A species is considered to possess a KEGG module if:
- It contains genes for >= 2/3 of the required steps
- For modules with alternative paths, only one must meet the 2/3 threshold
- Modules with <=3 steps require all steps present
- Module Abundance (Cellular Abundance): Computed as the sum of relative abundances of species possessing the module
- Gut-Brain Modules (GBMs)
- 56 microbial pathways involved in the synthesis/degradation of neuroactive compounds (e.g., GABA, dopamine)
- Pathways defined using KO, TIGRFAM, and EggNOG orthologs
- Same 2/3-rule applies for species inclusion
- Gut Metabolic Modules (GMMs)
- 103 conserved gut microbiome pathways (e.g., SCFA production, bile acid metabolism)
- Module completion and quantification rules follow KEGG module logic
Cellular Abundance vs. Relative Abundance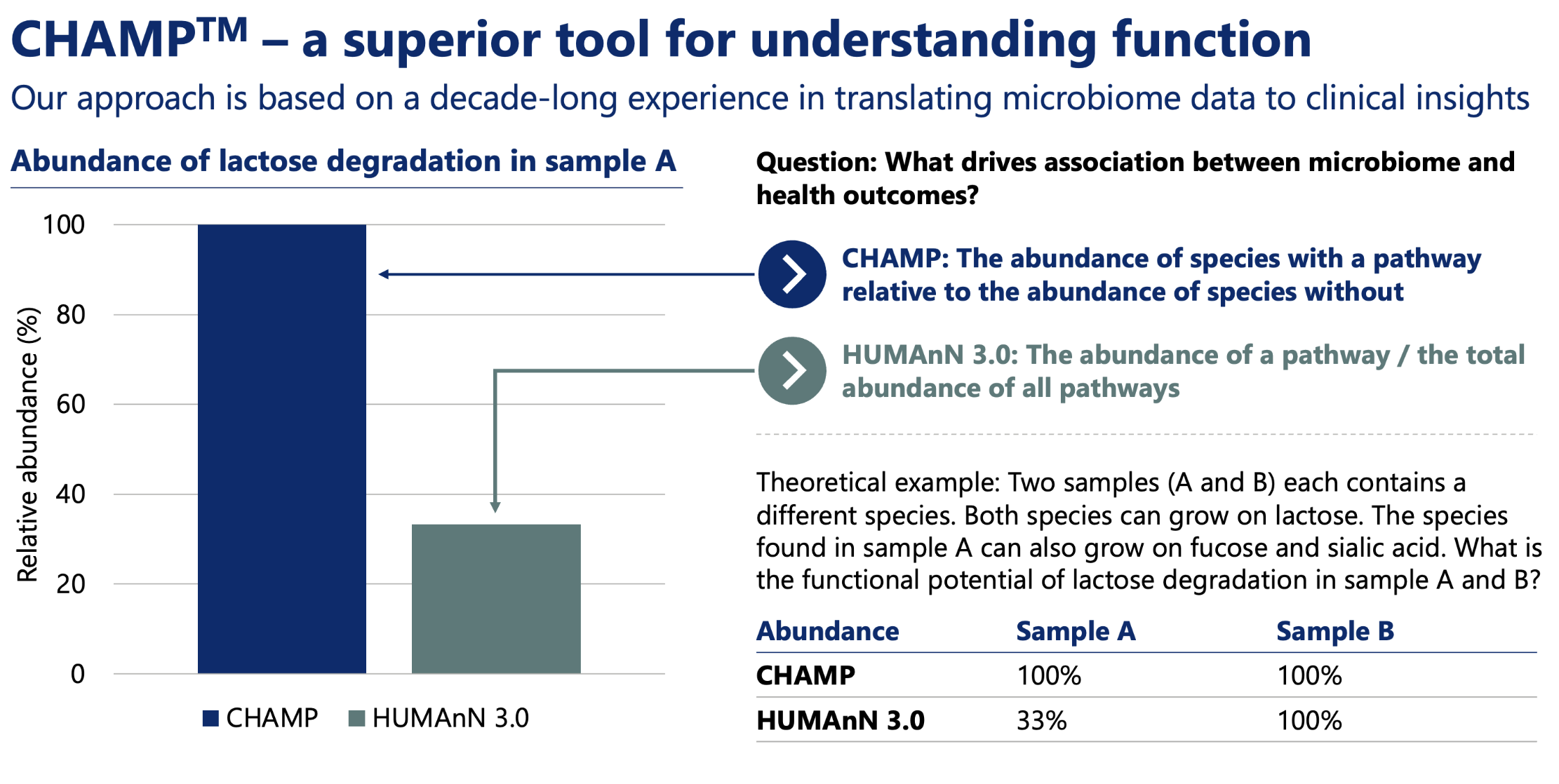
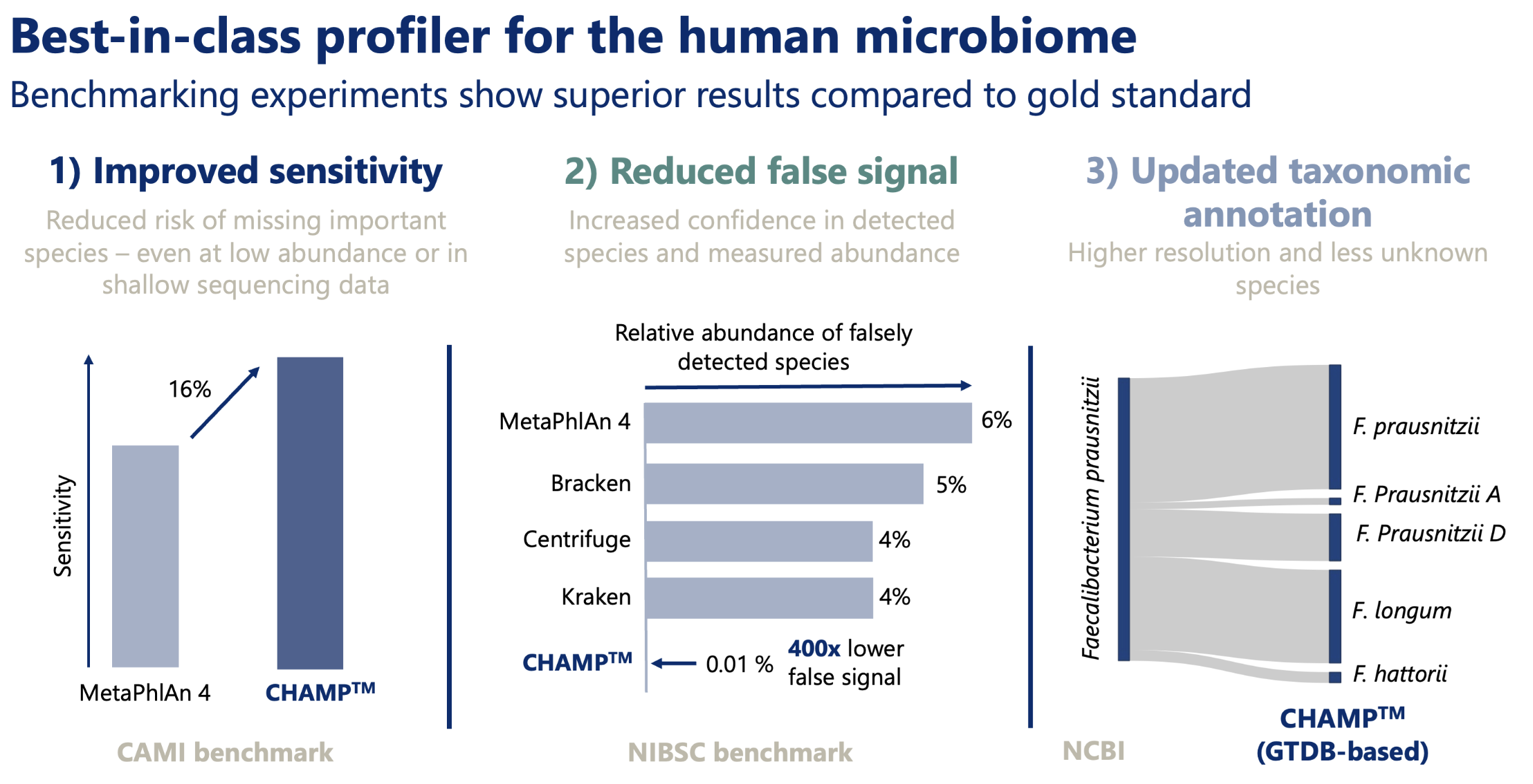
CHAMP Benchmarking
CHAMP demonstrates best-in-class performance using both CAMI and NIBSC benchmarks against the best and most widely used profiling pipelines in the field.- Compared to MetaPhlAn4, CHAMP showed 16% greater sensitivity (recall) across different human body sites and showed an astounding 400 times lower false signal in the NIBSC mock community benchmark compared to state-of-the-art profilers (MetaPhlAn4, Centrifuge, Kraken, and Bracken). This means that when CHAMP detects something, you can trust it’s there.
- CHAMP uses the latest GTDB annotation which includes more rare species and, compared to NCBI, often classifies sub-species as species in their own right.
- Read the benchmark whitepaper here: CHAMP Benchmark Whitepaper
Detailed Methods Summary
Gene Catalog and Species Definition
Gene Catalog and Species Definition
The Clinical Microbiomics Human Microbiome Reference HMR05 gene catalog is derived primarily from high-quality (HQ) prokaryotic MAGs (metagenome-assembled genomes) identified from 30,382 human microbiome samples collected from nine distinct human body sites: gut (n = 19,296), oral (n = 4994), skin (n = 4306), urine (n = 934), small intestine (n = 780), nasopharyngeal (n = 422), vaginal (n = 367), airway (n = 108), and milk (n = 100). This includes MAGs available from UHGG (89,262 MAGs, PMID: 32690973) and from ELGG (25,303 MAGs, PMID: 36050292). 11% (n = 3317) of the samples were not publicly available. In addition, genome assemblies from NCBI and PATRIC were added to capture otherwise missing species of interest (human-associated pathogens, probiotics, food ingredients, and species relevant for benchmarking). Human-relevant eukaryotic species were manually identified from various sources, including an analysis of gut fungal species (PMID: 29178920), publicly available lists of pathogens, the eukaryotes profiled by MetaPhlAn 4 (PMID: 36823356), and various species relevant for benchmarking, resulting in 2740 genomes representing 244 species.For MAGs not obtained from publicly available MAG collections, reads were host-filtered, trimmed, and assembled into contigs with Megahit (v. 1.2.9, PMID: 25609793) or metaSPAdes (v. 3.15.5, PMID: 28298430), and then binned using VAMB (v. 3.0.6, PMID: 33398153). MAGs were considered high-quality if they had > 90% completeness and < 5% contamination based on CheckM2 (v. 2022-07-19) and passed the GUNC chimerism test (v. 1.0.5, PMID: 34120611). All MAGs were taxonomically annotated using GTDB-Tk (v. 2.3.0, PMID: 36218463) with GTDB database version r214 (PMID: 34520557). To combine MAGs from multiple VAMB batches and MAG collections, MAGs annotated to the same species were merged into species clusters. MAGs without GTDB-Tk species-level annotations were merged with each other or with existing species clusters at 95% ANI (dRep, PMID: 28742071; FastANI, PMID: 30504855). This resulted in 6567 prokaryotic species clusters, 10% of which were unannotated at species level. To derive a pan-genome catalog for each species, we used a three-step clustering approach. First, genes were clustered with MMseqs2 (v. 14, PMID: 29959318) with 98% identity and 90% bi-directional coverage. Second, the representatives from the first iteration were clustered with MMseqs2 with 95% identity and 90% bi-directional coverage. Representatives of the second iteration were chosen as the ones with highest cardinality from the first iteration. Third, the second iteration representatives were clustered with cd-hit (cd-hit-est, v. 4.8.1, PMID: 23060610) with 95% identity and 90% coverage of the shorter sequence. Genes shorter than 100 bp or with species prevalence < 1% were discarded.For prokaryotes and eukaryotes separately, the entire set of pangenomes were then clustered with MMseqs2 with 97% identity and 90% bi-directional coverage to obtain between-species clusters. The pan-genomes from prokaryotic (n = 6567) and eukaryotic (n = 244) species were merged into a final gene catalog of 25,761,278 genes. To enable quantification of each species in the database, up to 250 signature genes were selected for each species based on core genes (> 60% prevalence in species MAGs) with a length > 200 bp and < 20 kbp. Furthermore, signature genes were required to be species-unique, with no alignments of 100 bp with > 97% sequence-identity to other genes in the catalog; however, if fewer than 20 genes meeting this criteria were available for a species, then genes with segments > 200 bp without alignments to other genes were used, and non-unique segments of these genes were masked.
Sequencing data preprocessing
Sequencing data preprocessing
Raw FASTQ files were filtered to remove human genomic contamination by discarding read pairs in which either read mapped to the human reference genome GRCh38.p14 with Bowtie2 (v. 2.4.2, Langmead and Salzberg 2012, PMID: 22388286) in local alignment mode. Reads were then trimmed to remove adapters and bases with a Phred score below 30 using AdapterRemoval (v. 2.3.1, Schubert et al. 2016, PMID: 26868221). Trimmed, host-filtered read pairs with both lengths > 100 bp, defined as high-quality non-host (HQNH) reads, were retained
Mapping reads to the gene catalog
Mapping reads to the gene catalog
HQNH reads were mapped to the gene catalog using BWA mem (v. 0.7.17) (Li and Durbin 2009, PMID: 19451168). An individual read was considered uniquely mapped to a gene if the MAPQ was >= 20 and the read aligned with >= 95 % identity over >= 100 bp. However, if > 10 bases of the read did not align to the gene or extend beyond the gene, the read was considered unmapped. Reads meeting the alignment length and identity criteria but not the MAPQ threshold were considered multi-mapped.Each read pair was counted as either 1) uniquely mapped to a specific gene, if one or both individual reads were uniquely mapped to a gene, or 2) multi-mapped, if neither read was uniquely mapped, and at least one was multi-mapped, or 3) unmapped, if both individual reads were unmapped. If the two reads were each uniquely mapped to a different gene, the gene mapped by read 1 was counted but not the gene mapped by read 2. A gene count table was created with the number of uniquely mapped read pairs for each gene.
Read Count Adjustment and Reporting
Read Count Adjustment and Reporting
The sample-specific removal of outlier genes for abundance estimation means that the effective genome length of a given species often varies from sample to sample. Thus, the raw species read counts are not directly proportional to the estimated species abundance and thus cannot be readily compared using standard count-based statistical methods. Therefore, the species read counts were adjusted to be proportional to species abundances with the following procedure: First, each species abundance was multiplied by the sum of its signature gene effective gene lengths (with all signature genes included, even those that were removed as outliers). Second, these values were scaled to sum to the total number of reads mapping to signature genes. Finally, these values were rounded to integers such that the rounded/unrounded discrepancy was minimized while retaining the total number of reads.
Species relative abundance calculation
Species relative abundance calculation
The expected read counts for signature genes in each species in each sample were modeled with a negative binomial distribution as follows. First, if >=50 of the signature genes for a species had non-zero read counts and >=99% of genes were expected to have non-zero read counts given the total read count for that species (1-(((n_genes-1)/n_genes)^n_reads)>=0.99), then signature genes with zero reads were ignored in that sample. Second, the expected 99% quantile (between 0.5% and 99.5%) of read counts were calculated for each gene based on a negative binomial distribution with a mean proportional to the effective gene length (accounting for read length and mapping alignment criteria) and dispersion defined as log2(effective gene length). The abundance of each species was then calculated as the mean read count normalised by effective gene length based on reads mapping to signature genes with observed read counts within the expected 99% quantile. Species abundances were set to zero if less than 5 genes with non-zero read counts were within the 99% quantile. Furthermore, species with <66% of genes with non-zero read count within the 99% quantile were set to zero, unless the median abundance of signature genes was non-zero, in which case the median gene-length-corrected abundance of non-zero genes was used. Abundances were then normalized sample-wise such that the total abundance of all species sums to 100 %.
Functional annotation and profiling
Functional annotation and profiling
EggNOG-mapper (v. 2.1.7, Diamond mode, Cantalapiedra et al. 2021, PMID: 34597405) was used to map prokaryotic genes in the gene catalog to the EggNOG orthologous groups database (v. 5.0, Huerta-Cepas et al. 2019, PMID: 30418610) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) database. Eukaryotic genes were annotated using KofamScan (PMID: 31742321). Functional potential profiles based on KOs were calculated as the proportion of the total gene abundance that mapped to a given KO.Kyoto Encyclopedia of Genes and Genomes (KEGG) modules (v. 78.2) (Kanehisa and Goto 2000, PMID: 10592173) are defined as a set of KOs that enable a specific function or pathway. Functional potential profiles based on KEGG modules were generated from the species profiles. For this, we identified the set of species associated to each of the KEGG modules by following three criteria: 1) a species was associated to a KEGG module if it included at least 2/3 of the genes encoding the proteins/enzymes needed to complete the functionality of the module, 2) if a module had alternative reaction paths, only one of these was required to be 2/3 complete, 3) for modules with three or fewer steps, all steps were required to be comprised in the given species. KEGG module profiles based on counts, relative abundances, rarefied counts and rarefied relative abundances were then calculated by adding, respectively, the number of counts, relative abundances, rarefied counts and rarefied relative abundances from each species associated to a given KEGG module.
Gut-Brain Modules (GBM)
Gut-Brain Modules (GBM)
New AccordionThe gut-brain modules (GBMs) are a set of 56 microbial pathways for metabolizing neuroactive compounds (molecules that have the potential to interact with the human nervous system). Each GBM corresponds to a single neuroactive compound synthesis or degradation process by members of the gut microbiota and is defined as a series of enzymatic steps represented by ortholog group identifiers (KEGG, TIGRFAM, and eggNOG version 3.0 orthology databases in order of preference) (Valles-Colomer et al. 2016, PMID: 30718848). We consider a species to contain a given module if the species includes genes annotated to at least 2/3 of the steps needed to complete the functionality of the module. If a module has alternative reaction paths, only one of these is required to be 2/3 complete. For modules with three or fewer steps, all steps are required to be comprised in the species.
Gut Metabolic Modules (GMM)
Gut Metabolic Modules (GMM)
Gut Metabolic Modules (GMMs) are a set of 103 conserved human gut metabolic pathways, each defined as a series of enzymatic steps represented by Kyoto Encyclopedia of Genes and Genomes orthology (KO) identifiers (Vieira-Silva et al. 2016, PMID: 27573110). Functional potential profiles based on GMMs were generated from the species profiles. For this, we identified the set of species associated to each of the GMMs by following three criteria: 1) a species was associated to a GMM if it included at least 2/3 of the genes encoding the proteins/enzymes needed to complete the functionality of the module, 2) if a module had alternative reaction paths, only one of these was required to be 2/3 complete, 3) for modules with three or fewer steps, all steps were required to be comprised in the given species. GMM profiles based on counts, relative abundances, rarefied counts and rarefied relative abundances were then calculated by adding, respectively, the number of counts, relative abundances, rarefied counts and rarefied relative abundances from each species associated to a given GMM.