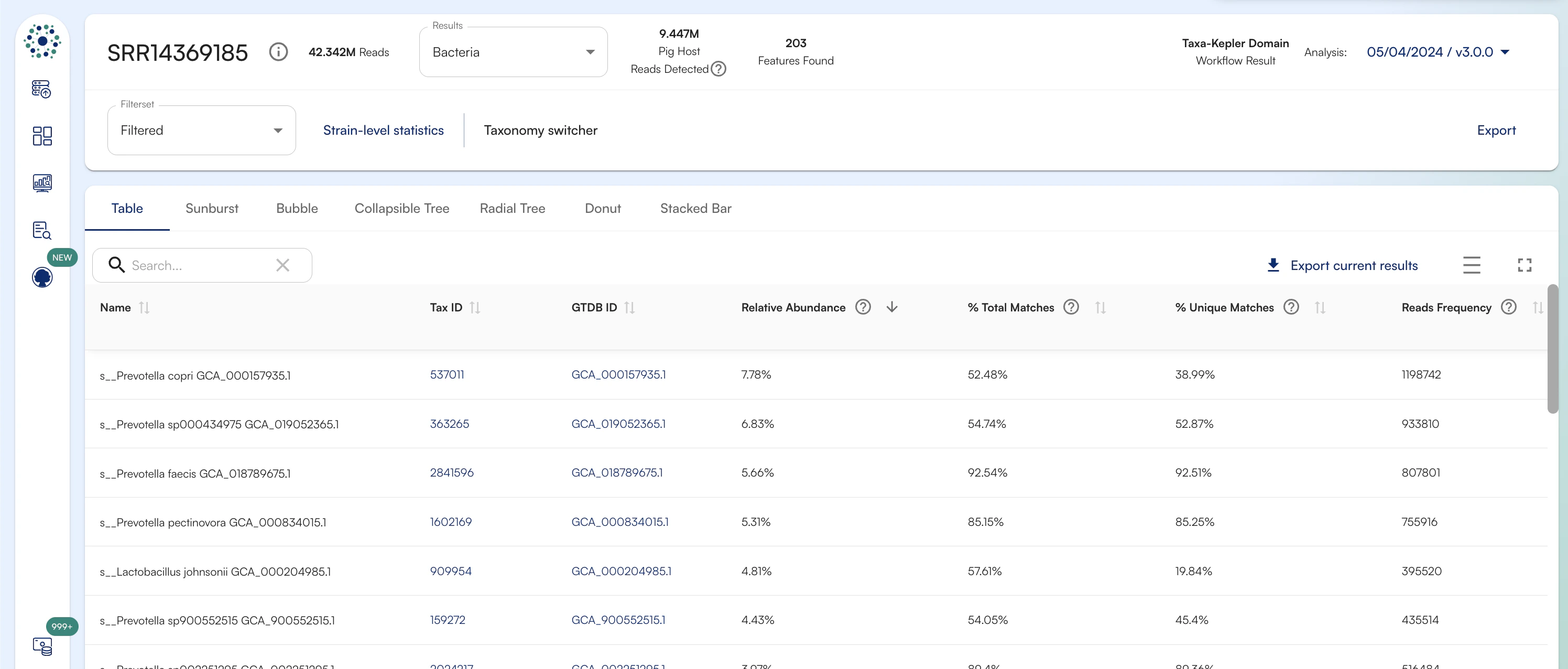
% Total vs. Unique Matches% Total Matches is the number of shared+unique kmer matches identified in the sample divided by the total number of pre-calculated shared+unique matches available in the reference database. Shared kmers are shared with similar organisms of the same lineage in the phylogenetic tree, which makes this a useful metric for approximating gene coverage, or for assessing the likelihood of a detected taxon or gene actually being present.% Unique Matches is the number of unique kmers found in the sample that are unique to the organism divided by the total number of pre-calculated unique kmers available for the organism in the database. If unique coverage is very low for an organism, it could mean a couple of things:
- For strains, this could indicate that the exact strain in our reference database is not a perfect match to the strain in the sample. However, if you also see a high percent TOTAL coverage for this same strain, this is a good indication that a near taxonomic neighbor of the reference strain is in your sample.
- For some organisms that have high representation in sequencing space (E. coli, for example), there may not be many unique areas in the genome available for strain level identification. As more and more similar genomes are added to the database, we trade-off the ability to discriminate between those that are highly similar to each other. This will cause the percent unique coverage to be low.
This metric is not recommended for downstream comparative analysis.This metric is used by the Filtered setting to rule out likely false positive calls.A common use of % Total Matches is for comparative analysis of antibiotic resistance genes or virulence factors, as these databases are gene-based rather than organism-based. The number of total k-mers identified enables a meaningful in understanding how well the gene has been covered by the reads in the sample.Use the % Total Matches metric to compare how coverage changes between samples, and use the Relative Abundance metric to compare how the composition of marker genes changes between samples.