KEPLER Host-Agnostic Taxonomic Profiling
KEPLER is a novel taxonomic profiling pipeline engineered to extract optimal value from metagenomic data. Combining the sensitivity of K-mer exact-matching and the precision and accuracy of probabilistic alignment, Kepler achieves robust identification and enumeration of microbial taxa spanning bacteria, viruses, fungi, and protists. Significantly, Kepler's accuracy and precision relies upon a meticulously curated biomarker database, where over 30,000 species across Bacteria, Viruses, Protists and Fungi are arranged in a phylogenetic tree-like structure.
The core of KEPLER's technology is patented in both the US Patent office (US10108778B2, US20200294628A1) and European Patent Office (ES2899879T3).
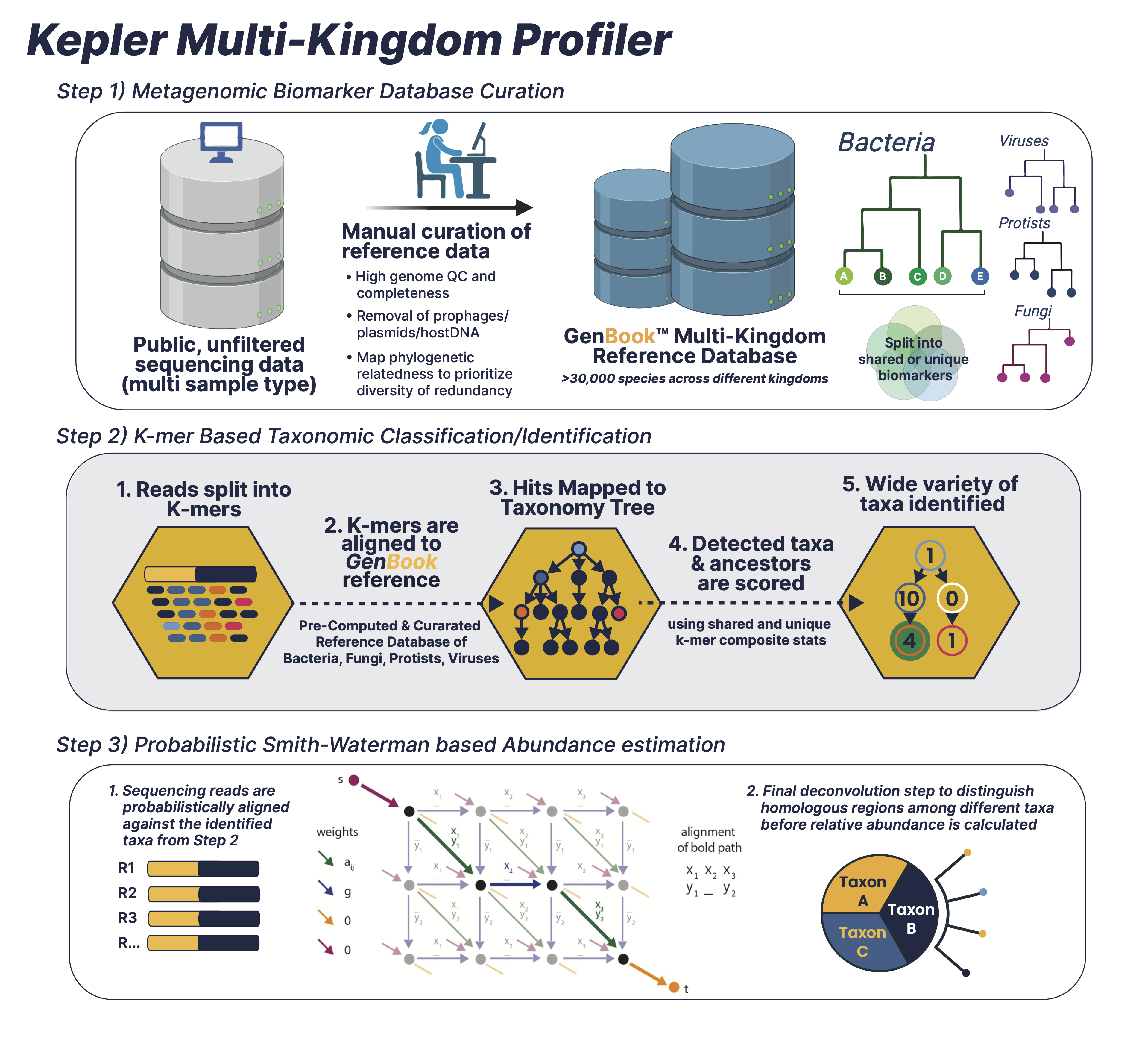
How does KEPLER work?
The patented KEPLER multi-kingdom taxonomic profiler is a comprehensive algorithm consisting of three interwoven pipelines:
1. Pre-computational stage for curating and building a comprehensive biomarker database (GenBook™)
The first iteration of database curation involves the selection of high quality microbial genomes based on high completeness/low contamination ratio, quality of genome assembly (genomes with high fragmentation/# of contigs discarded when better representatives for that clade are present), and prioritization of intra-species diversity instead of redundancy. Once the library of high quality genome assemblies are selected, the genome assemblies are then scrubbed clean of low complexity sequences, prophages, plasmids, and human/mouse contaminated regions to maximize for taxonomically informative signal to noise ratio. The final database (GenBook™) includes >30,000 species encompassing multiple microbial kingdoms including Bacteria, Viruses, Protists, Fungi, and Phages. Next, the second iteration of database building involves splitting the curated and cleaned collection of microbial genomes into n-mers of variable length. The n-mers are then categorized as either shared or unique biomarkers across individual genomes, which is facilitated by a phylogenetic tree-like data structure. The tree backbone represents shared genomic biomarkers between different taxa, while the tree leaves are individual microbial genomes with unique biomarkers.
2. K-mer based Taxonomic classification/Identification
This first comparator phase splits the millions of sequencing reads for each sample into k-mer sets. It then looks for exact matches between sample k-mers and reference biomarkers that are dispersed across the different phylogenetic branches and leaves of the GenBook™ database. . By focusing on small genomic regions of large differential informational content, this phase eliminates 99% of genomes which are almost certainly not in the metagenomic sample and generates a short list of nearest reference strains. This highly efficient and accurate classification readily admits sub-species taxonomy to an ANI divergence less than 0.003. Classification sensitivity and accuracy is maintained through biomarker aggregation statistics, coverage depth estimation, and abundance estimation–but with an inhomogeneous variance that is parsed into Step (3).
3. Probabilistic Smith-Waterman based Abundance Estimation and Classification Refinement
The second comparator phase uses an edit distance-scoring based probabilistic Smith-Waterman algorithm [1] to compare sequencing reads with a reference set of identified microbial taxa from Step (2). This involves a more computationally-intensive processing of the remaining 1% of strains to eliminate any false positives and achieve abundance estimations which are more homogeneous, more accurate, and have significantly less variance.
Given an example scenario of 10 reads–where reads 1-8 belong to strain A, read 9 belongs to strain B, and read 10 belongs to both strain A and strain B with equal alignment score–KEPLER will probabilistically split read 10’s abundance into strain A with an abundance of 8.89 and strain B with an abundance of 1.11. Given a more complicated metagenomic sample scenario, contested reads are apportioned according to implied priors from uncontested reads. An iterative Maximum Likelihood Estimation algorithm is then converged to the final read abundance estimates. To summarize, overall abundance precision and classification accuracy is achieved by running the comparators in sequence, scoring the entire read probabilistically against the reference set, and distinguishing homologous regions.
KEPLER Benchmarking
The performance of KEPLER on benchmark datasets, as highlighted in our benchmark whitepaper, demonstrates its superiority in species-level identification and functional annotation. KEPLER outperformed other tools in detecting microbial species, especially in complex environments like low-biomass samples. It showed enhanced accuracy and precision in taxonomic classification across a range of sample types.
Key findings from the benchmarking include:
- Higher Sensitivity: KEPLER was able to detect microbial species that other tools missed, particularly in datasets with lower microbial diversity.
- Improved Speed and Scalability: Compared to competing tools, KEPLER processed larger datasets more quickly, making it suitable for large-scale studies.
- Superior Functional Annotation: The tool provided more detailed functional profiles, linking microbial presence to potential biological functions more effectively than other systems.
To learn more about KEPLER and its benchmarking, please download the whitepaper from here.
Cited by:
The papers mentioned below can be cited for referencing to our taxonomic profiling method.
-
https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2023.1165980/full
-
https://www.cell.com/cell-host-microbe/pdfExtended/S1931-3128(19)30158-1
You can find additional studies at our literature page.
FAQ
To learn more about KELPER™ and its benchmarking, please download the KEPLER Benchmarking whitepaper https://cosmosidhub.com/kepler-host-agnostic-microbiome-profiler/ .
-
I have samples I ran previously on CosmosID-HUB and I would like to rerun them with KEPLER, how do I do that?
- Please contact [email protected] for more information on how to re-profile your existing data on CosmosID-HUB with KEPLER.
-
For upcoming/future Bioinformatics services project delivery, will I automatically get taxonomic profiling results through KEPLER?
- Yes, the Bioinformatics services team will deliver your data on KEPLER going forward. If you would like your old data re-profiled using KEPLER, please contact [email protected].
-
Will my KEPLER profiling results differ from the current taxa workflow results?
- Yes, some differences are expected due to updates of the algorithm, database and taxonomy.
-
Will KEPLER provide taxonomic profiling data with GTDB taxonomy?
- Yes, KEPLER uses GTDB taxonomy for providing lineage information on identified Bacteria and Archaea. To learn more about the differences between GTDB taxonomy and NCBI taxonomy for Bacteria and Archaea, please visit GTDB and NCBI taxonomy comparison
Updated 3 days ago